A Unified Quantile Framework for Transcriptome-Wide Association Analysis
Methodology
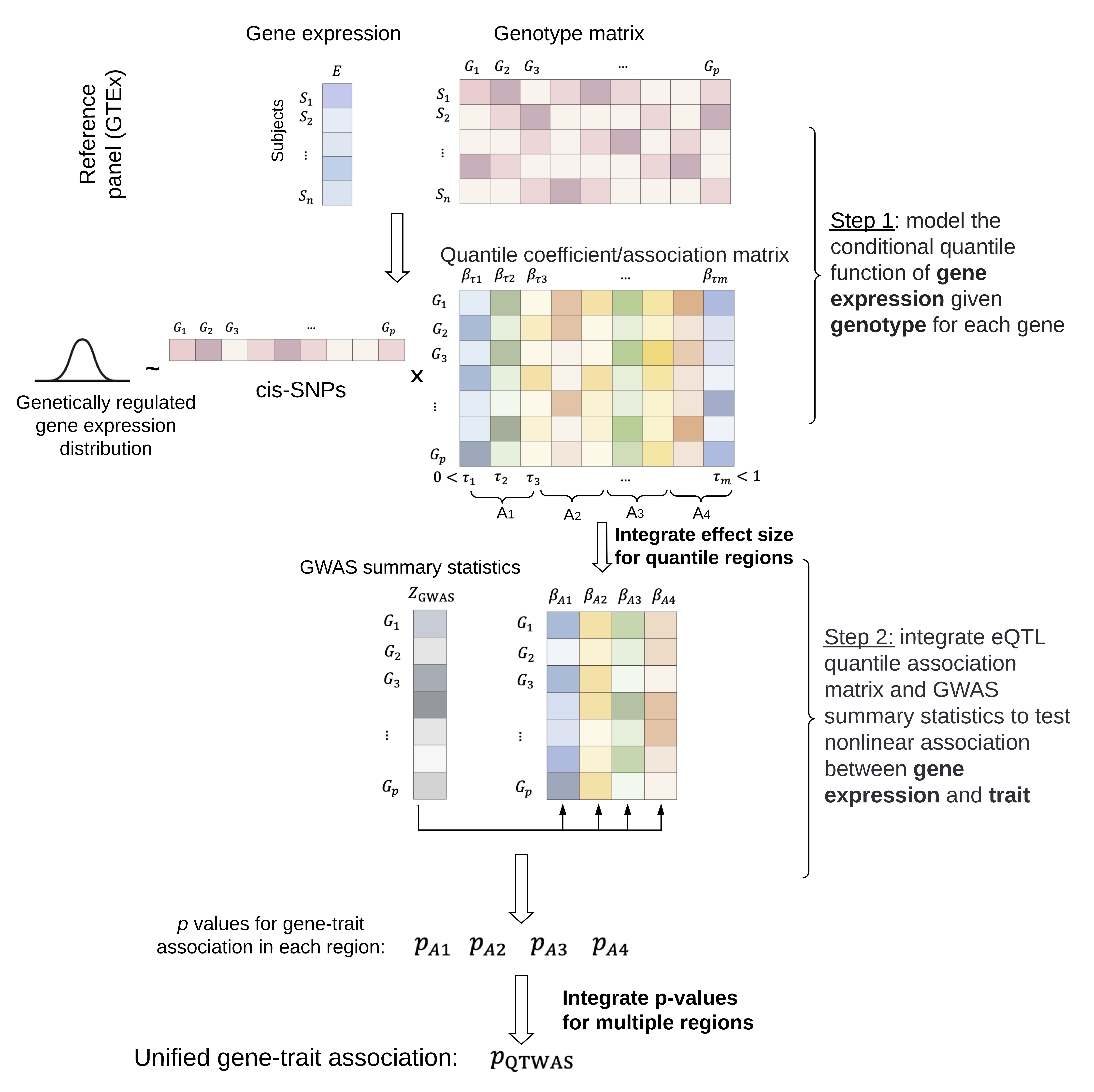
- Please see the manuscript “A Unified Quantile Framework reveals nonlinear heterogeneous transcriptome-wide associations” for more details.
Software
We developed software tools based on R: QTWAS for training gene expression model on based on GTEx data. Please see the Github page for more details.
Models
We provide pre-trained QTWAS models per gene for 49 tissues in GTEx v8. R scores for evaluating imputation accuracy are also provided for each model.
- Download all the QTWAS models here. In each database file, we provide the information for:
- estimated beta at selected SNPs, indexed by rsid and gene ensemble ID. interval represents the quantile region of gene expression, i.e.,
.
- covariance matrix of SNPs in the imputation models, indexed by rsid and gene ensemble ID.
Code to extract the information:
driver <- dbDriver('SQLite') conn <- dbConnect(drv = driver, file.name) #use the file you want to extract for file.name mytable.beta <- dbReadTable(conn,"beta") mytable.cov_mat <- dbReadTable(conn,"cov_mat") dbDisconnect(conn) - estimated beta at selected SNPs, indexed by rsid and gene ensemble ID. interval represents the quantile region of gene expression, i.e.,
- Download R scores for each model here
Results
Ten psychiatric/disorder diseases
Here we provide QTWAS results on the summary statistics from ten GWAS studies on brain disorders, including five neuropsychiatric traits: schizophrenia (SCZ1), attention-deficit/hyperactivity disorder (ADHD2), bipolar disorder (BD3), autism spectrum disorder (ASD4) and major depressive disorder (MDD5); and four neurodegenerative traits: Alzheimer’s disease (AD_Kunkle6 and AD_Jansen7), Parkinson’s disease (PD8), multiple sclerosis (MS9) and amyotrophic lateral sclerosis (ALS10).
-
Download Manhattan plots per triat (multi-tissue results based on 13 brain tissues)
-
Download QQ plots per tissue per trait for all 49 tissues
-
Download Significant genes per tissue per trait for all 49 tissues
-
Download QTWAS p values for all genes per tissues per trait for all 49 tissues
We also developed a Shiny app to visualize the results: tianyingw.shinyapps.io/QTWAS/.
797 UK Biobank traits
We also provide QTWAS results on 797 UK Biobank continuous phenotypes with their summary statistics on 28 million imputed variants. We provide phenome-wide results for genes, genome-wide gene-based results for each trait with respect to all 49 tissues separately.
- Download QTWAS p values for all genes per tissues per trait for all 49 tissues
References
-
Pardiiñas, A. F., Holmans, P., Pocklington, A. J., Escott-Price, V., Ripke, S., Carrera, N., Legge, S. E., Bishop, S., Cameron, D., Hamshere, M. L., et al. (2018). Common schizophrenia alleles are enriched in mutation-intolerant genes and in regions under strong background selection. Nature genetics, 50, 381–389. ↩
-
Demontis, D., Walters, R. K., Martin, J., Mattheisen, M., Als, T. D., Agerbo, E., Baldursson, G., Belliveau, R., Bybjerg-Grauholm, J., Bækvad-Hansen, M., et al. (2019). Discovery of the first genome-wide significant risk loci for attention deficit/hyperactivity disorder. Nature genetics, 51, 63–75. ↩
-
Sarlus, H., Heneka, M. T., et al. (2017). Microglia in alzheimer’s disease. The Journal of clinical investigation, 127, 3240–3249. ↩
-
Grove, J., Ripke, S., Als, T. D., Mattheisen, M., Walters, R. K., Won, H., Pallesen, J., Agerbo, E., Andreassen, O. A., Anney, R., et al. (2019). Identification of common genetic risk variants for autism spectrum disorder. Nature genetics, 51, 431–444. ↩
-
Howard, D. M., Adams, M. J., Clarke, T.-K., Hafferty, J. D., Gibson, J., Shirali, M., Cole- man, J. R., Hagenaars, S. P., Ward, J., Wigmore, E. M., et al. (2019). Genome-wide meta-analysis of depression identifies 102 independent variants and highlights the impor- tance of the prefrontal brain regions. Nature neuroscience, 22, 343–352. ↩
-
Kunkle, B. W., Grenier-Boley, B., Sims, R., Bis, J. C., Damotte, V., Naj, A. C., Boland, A., Vronskaya, M., Van Der Lee, S. J., Amlie-Wolf, A., et al. (2019). Genetic meta-analysis of diagnosed alzheimer’s disease identifies new risk loci and implicates aβ, tau, immunity and lipid processing. Nature genetics, 51, 414–430. ↩
-
Jansen, I. E., Savage, J. E., Watanabe, K., Bryois, J., Williams, D. M., Steinberg, S., Sealock, J., Karlsson, I. K., Ha ̈gg, S., Athanasiu, L., et al. (2019). Genome-wide meta- analysis identifies new loci and functional pathways influencing alzheimer’s disease risk. Nature genetics, 51, 404–413. ↩
-
Nalls, M. A., Blauwendraat, C., Vallerga, C. L., Heilbron, K., Bandres-Ciga, S., Chang, D., Tan, M., Kia, D. A., Noyce, A. J., Xue, A., et al. (2019). Identification of novel risk loci, causal insights, and heritable risk for parkinson’s disease: a meta-analysis of genome-wide association studies. The Lancet Neurology, 18, 1091–1102. ↩
-
Andlauer, T. F., Buck, D., Antony, G., Bayas, A., Bechmann, L., Berthele, A., Chan, A., Gasperi, C., Gold, R., Graetz, C., et al. (2016). Novel multiple sclerosis susceptibility loci implicated in epigenetic regulation. Science advances, 2, e1501678. ↩
-
Van Rheenen, W., Shatunov, A., Dekker, A. M., McLaughlin, R. L., Diekstra, F. P., Pulit, S. L., Van Der Spek, R. A., Vo ̃sa, U., De Jong, S., Robinson, M. R., et al. (2016). Genome-wide association analyses identify new risk variants and the genetic architecture of amyotrophic lateral sclerosis. Nature genetics, 48, 1043–1048. ↩